N6-Methyladenosine (m6A)

m6A RNA Methylation and Demethylation Cycle. Credit: EpiGentek/Ferrufino E. (2017). Click to enlarge.
N6-methyladenosine, otherwise known as m6A, is the most abundant internal modification in mRNA. It entails methylation of the adenosine base at the nitrogen-6 position. Previous studies have demonstrated that m6A is a reversible and dynamic RNA modification that is integral to the regulation of RNA metabolism. Similar to DNA and histone modification, RNA methylationpatterns play an important biological function in the regulation of different cellular processes, such as metabolism, embryonic development, and stem cell self-renewal.
m6A is catalyzed by a methyltransferase complex which consists of “writer” proteins methyltransferase-like 3 (METTL3), methyltransferase-like 14 (METTL14), and Wilms tumor 1 associated protein (WTAP). METTL3 and METTL14 form a heterodimer in which METTL3 acts as a catalyst while METTL14 stabilizes METTL3 as it modifies the RNA molecule. WTAP then interacts with this heterodimer in order to localize the methylase complex to specific loci in the nucleus.

m6A demethylation is catalyzed by two “eraser” demethylases, fat mass and obesity-associated protein (FTO) and AlkB homolog 5 (ALKBH5). The demethylases catalyze m6A demethylation in an α-KG and Fe2+ dependent manner. YTHDF1, YTHDF2, YTDHF3, and YTHDC1, members of the YTH domain family of proteins, have been identified as m6A readers that recognize the modification and affect the pre-mRNA splicing as well as mRNA transport, stability, and translation.
High-throughput sequencing experiments have demonstrated that m6A modifications are not randomly distributed, but are instead enriched at specific transcript landmarks. Interestingly, m6A modifications are typically found clustered around stop codons, in 3’UTRs, and within long internal exons. The locations of these modifications imply a role in translational control. Furthermore, previous studies have shown that m6A modifications in mRNAs or non-coding RNAs play essential roles in various bioprocesses including embryonic stem cell maintenance and differentiation, meiotic progression, circadian rhythm, heat shock response, and neuronal function.
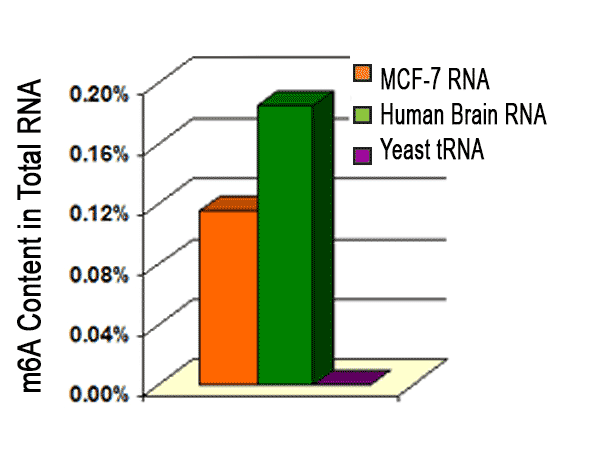 For absorbance-based quantitation of N6-methyladenosine in RNA in an ELISA-like format use the EpiQuik m6A RNA Methylation Quantification Kit (Colorimetric). This kit is ideal for direct detection of m6A RNA methylation status using total RNA isolated from any species. View our wide range of RNA methylation kitsand our m6A Antibody.
For absorbance-based quantitation of N6-methyladenosine in RNA in an ELISA-like format use the EpiQuik m6A RNA Methylation Quantification Kit (Colorimetric). This kit is ideal for direct detection of m6A RNA methylation status using total RNA isolated from any species. View our wide range of RNA methylation kitsand our m6A Antibody.

Self-renewal of glioblastoma stem cells is regulated by m6A RNA methylation. Credit: Cui C. et al. (2017).
m6A and Cancer
There have also been links between alteration in m6A levels and abnormal cellular differentiation states present in cancer. m6A modifications have been shown to play a role in leukemia, brain cancer, breast cancer, and lung cancer.
The key proteins for m6A modification, WTAP and FTO, have been shown to be highly expressed in acute myeloid leukemia (AML), AML-carrying NPM1 mutations and/or FLT3-ITD, which suggests that m6A modification in this subtype of AML is tightly and sophistically controlled by both writers and eraser regulators.
A study from the University of Chicago has demonstrated that FTO plays an oncogenic role in promoting leukemic cell transformation and AML cell survival and growth. In vitroforced expression of FTO significantly enhanced the viability and growth of human AML cells. An m6A-seq assay performed suggests that ASB2and RARAare two critical targets genes of FTO. FTO reduced m6A levels at the UTRs of ASB2and RARA, resulting in the downregulation of the RNA and protein level of both genes. This was further confirmed by the knockdown of FTO, which lead to an increase in the m6A levels on ASB2and RARA, while forced expression of FTO increased the m6A levels of ASB2and RARA.
In brain tumors, elevated expression of ALKBH5in glioblastomastem cells is an indication of a poor prognosis for the patient because ALKBH5 promotes tumorgenesis by enhancing FOXM1expression. Upon ALKBH5 knockdown, there is an increase in m6A modification on FOXM1nascent transcript resulting in a decrease in FOXM1expression. FOXM1 is a transcription factor that plays a critical role in the self-renewal and tumorgenesis of glioblastoma stem cell. Therefore, elevated ALKBH5expression leads to an increase in FOXM1expression, which in turn enhances glioblastoma stem cell self-renewal proliferation.
The knockdown of METTL3 and METTL14 has also been demonstrated to upregulate the expression levels of several oncogenes, including ADAM19, EPH3and KLF4, and downregulate the expression of tumor suppressor genes, such as CDKN2A. It also leads to a reduction in mRNA m6A levels, resulting in enhanced in vitrogrowth and self-renewal of glioblastoma stem cells, and an increase in the formation of brain tumors in vivo. In contrast, overexpression of METTL3 or treatment with an FTO inhibitor increased m6A levels in glioblastoma stem cells and suppressed their growth.

m6A regulatory protein regulation by hypoxia and its downstream effects in human breast cancer cells. Credit: EpiGentek/Ferrufino E. (2017). Click to enlarge.
In human breast cancer cells, hypoxia regulates m6A modification by increasing the demethylation of mRNA encoding core
pluripotency factors. Most solid tumors are in a hypoxic environment, which causes an increase in the expression of
HIF-1αand HIF-2α. As HIF activity increases, ALKBH5 activity increases, leading to transcriptional and
post-transcriptional changes in expression of genes that promote the specification and maintenance of breast cancer stem
cells. This is further confirmed by the knockdown of ALKBH5, which similar to its role in brain tumorigenesis, inhibits
tumor formation and causes a decrease in breast cancer stem cells. The upregulation of ALKBH5expression promotes
mRNA stability and expression of NANOG. This in turn promotes the formation of more breast cancer cells. NANOG, a
transcription factor involved in the self-renewal process of undifferentiated ESCs, is known to play a critical role in
the maintenance of cancer stem cells which are required for primary tumor formation and metasis.
METTL3has been observed to be upregulated in lung adenocarcinoma and is believed to play a role in growth, survival, and invasion of human lung cancer cells. The depletion of METTL3 leads to strong inhibition of cell growth, increase in cell apoptosis, and decrease in invasive ability of cancer cells. METTL3 promotes translation of certain mRNAs, such as EGFRand TAZ, which are involved in the regulation of cell proliferation and stem-cell renewal.
Targeting m6A Modification for Treatment Strategies
Overall, m6A modificationsand their regulatory proteins play a role in various types of cancers. Therefore, targeting m6A modification may serve as an effective treatment strategy. Because FTO has a functional role in the viability and growth of human AML cells, an FTO inhibitor may selectively inhibit FTO signaling and provide a therapeutic strategy to treat leukemia.
In glioblastoma, overexpression of METTL3or treatment with MA2, an inhibitor of FTO, has resulted in increased mRNA m6A levels and suppressed glioblastoma stem cell growth. Therefore, modifying m6A methylation could be a promising target for treating glioblastomas. METTL3 could also be a potential therapeutic target in lung cancer. METTL3 in lung cancer promotes oncogene expression, cancer cell growth, and survival. Therefore, the inhibition of METTL3 could suppress oncoprotein expression and prevent the growth and survival of cancer cells. Since m6A and its regulatory protein play a critical role in different types of cancers, the development of applicable inhibitors may provide more effective novel therapeutic strategies to treat cancer.
References:- Batista P.J. (2017). The RNA modification N6-methyladenosine and its implications in human disease. Genomic Proteomics Bioinformatics, 15: 154-163.
- Cui Q., et al. (2017). m6A RNA Methylation Regulates the Self-Renewal and Tumorgenesis of Glioblastoma Stem Cells. Cell Reports, 18: 2622-2634.
- Deng X., et al. (2018). Role of N6-methyladenosine modification in cancer. Current Opinions in Genetics and Development, 48: 1-7.
- Jaffrey S.R. and Kharas M.G. (2017).Emerging links between m6A and misregulated mRNA methylation in cancer. Genome Medicine, 9:2.
- Meyer K.D., et al. (2012). Comprehensive analysis of mRNA methylation reveals enrichment in 3’UTRs and near stop codons. Cell, 149: 1635-1645.
- Li Z., et al. (2017). FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N6-methyladenosine RNA Demethylase. Cancer Cell, 31: 127-141.
- Lin S., et al. (2016). The m6A Methyltransferase METTL3 Promotes Translation in Human Cancer Cells. Molecular Cell, 62: 335-345.
- Roundtree I.A., et al. (2017). Dynamic RNA modification in gene expression regulation. Cell. 169: 1187-1200.
- Wang S., et al. (2017). Role of RNA methylation by means of N6-methyladenosine (m6A) in human cancer. Cancer Letters, 408: 112-120.
- Wu B., et al. (2017). Readers, writers and erasers of of N6-methylated adenosine modification. Current Opinions in Structural Biology, 147: 67-76.
- Zhang C., et al. (2016). Hypoxia Induces the Breast Cancer Stem Cell Phenotype by HIF-dependent and ALKBH5-mediated m6A-demthylation of NANOG mRNA. PNAS, E2047-2056.