FFPE chromogenic IHC protocol Use this workflow for formalin-fixed paraffin-embedded tissue stained with HRP/DAB or another chromogenic detection system. Use this format when: Evaluating protein expression or localization in FFPE tissue sections by brightfield microscopy.Optimize first: Antigen retrieval buffer, retrieval time, primary antibody dilution, blocking, detection chemistry, and hematoxylin counterstain. 1 Bake and deparaffinize slidesBake 4-5 um FFPE sections at 60 degrees C for 30-60 minutes. Deparaffinize in xylene, 2 changes for 5 minutes each. Rehydrate through 100 percent, 95 percent, and 70 percent ethanol, 2-3 minutes each, then rinse in water. 2 Perform antigen retrievalPlace slides in citrate buffer pH 6.0 or Tris-EDTA buffer pH 9.0. Heat near boiling for 10-20 minutes using a pressure cooker, microwave, steamer, or water bath. Cool slides in buffer for 20 minutes, then rinse in TBS or PBS. 3 Block endogenous activityFor HRP detection, incubate slides in 3 percent hydrogen peroxide for 10 minutes to quench endogenous peroxidase. Rinse 3 times in buffer. 4 Block nonspecific bindingApply protein block, 5 percent normal serum, or 1-5 percent BSA for 20-60 minutes at room temperature. Drain but do not rinse if the detection system recommends leaving blocking protein on the section. 5 Add primary antibodyDilute 5-Methylcytosine (5-mC) Monoclonal Antibody [33D3] in antibody diluent. Start with the datasheet-recommended IHC dilution or test 1:50, 1:100, 1:250, and 1:500. Apply 100-200 uL per section and incubate 1 hour at room temperature or overnight at 4 degrees C in a humidified chamber. 6 Wash after primary antibodyWash 3 times for 5 minutes each in TBS-T or PBS-T. Keep slides covered with buffer and do not let tissue dry. 7 Apply secondary or polymer detectionApply HRP polymer, biotinylated secondary plus streptavidin-HRP, or another detection reagent according to the system instructions. Typical incubation is 20-30 minutes at room temperature. Wash 3 times. 8 Develop chromogenApply DAB or other chromogen for 1-10 minutes while monitoring signal under a microscope. Stop development by rinsing in water. 9 Counterstain and mountCounterstain with hematoxylin for 15 seconds to 2 minutes, blue in running tap water or bluing reagent, dehydrate through ethanol, clear in xylene, and mount with permanent mounting medium. Retrieval optimizationIf signal is weak, compare pH 6 and pH 9 retrieval before dramatically increasing antibody concentration.Negative controlUse no-primary, isotype control when appropriate, and known negative tissue to evaluate nonspecific staining.
Frozen tissue IHC protocol Use this workflow for frozen sections when antigen preservation is more important than FFPE morphology. Use this format when: The epitope is fixation-sensitive, lipid-rich tissue is used, or frozen-section staining is preferred.Optimize first: Fixation method, section thickness, drying time, blocking, primary antibody dilution, and tissue autofluorescence or endogenous enzyme activity. 1 Prepare frozen sectionsCut 5-10 um cryosections and mount onto charged slides. Air dry 10-30 minutes at room temperature. Store slides cold if staining later. 2 Fix sectionsFix with cold acetone for 10 minutes at -20 degrees C, cold methanol for 5-10 minutes, or 4 percent paraformaldehyde for 10-15 minutes depending on the target. Rinse gently in PBS or TBS. 3 Block endogenous activity if neededFor HRP-based detection, quench endogenous peroxidase with 0.3-3 percent hydrogen peroxide for 10 minutes. For biotin-based detection, consider avidin/biotin blocking when tissue has high endogenous biotin. 4 Block nonspecific bindingBlock 20-60 minutes with 5 percent normal serum or 1-5 percent BSA in PBS/TBS. Use serum from the secondary antibody host species when practical. 5 Add primary antibodyDilute 5-Methylcytosine (5-mC) Monoclonal Antibody [33D3] in antibody diluent. Start with the datasheet-recommended frozen IHC dilution or test 1:50 to 1:500. Apply 100-200 uL per section and incubate 1 hour at room temperature or overnight at 4 degrees C. 6 Wash and detectWash 3 times for 5 minutes. Apply secondary antibody, polymer detection, or fluorescent secondary antibody according to the detection system. Incubate 30-60 minutes. 7 Develop or mountFor chromogenic detection, apply substrate until signal develops, rinse, counterstain, and mount. For fluorescent detection, protect from light, counterstain with DAPI if needed, and mount with antifade medium. MorphologyFrozen sections usually preserve antigenicity better but morphology can be less crisp than FFPE tissue.Section adhesionUse charged slides and avoid harsh washing if sections lift from the slide.
Fluorescent IHC protocol Use this workflow for fluorescent staining of tissue sections when colocalization or multiplex imaging is needed. Use this format when: Tissue localization is needed with fluorescent readout, multiple markers, or confocal microscopy.Optimize first: Autofluorescence control, antigen retrieval, fluorophore choice, antibody host species, and mounting medium. 1 Prepare and retrieve tissueFor FFPE tissue, deparaffinize, rehydrate, and perform antigen retrieval. For frozen tissue, fix according to target requirements. Rinse in PBS or TBS. 2 Reduce autofluorescenceTreat autofluorescent tissues with a compatible quenching reagent when needed. Avoid quenchers that interfere with the planned fluorophores. 3 Permeabilize and blockUse 0.1-0.3 percent Triton X-100 for intracellular access when compatible. Block with 5 percent normal serum or 1-5 percent BSA for 30-60 minutes. 4 Add primary antibodyDilute 5-Methylcytosine (5-mC) Monoclonal Antibody [33D3] in blocking buffer. Start with the datasheet-recommended IHC/IF dilution or test 1:100, 1:250, and 1:500. Apply 100-200 uL per section and incubate 1-2 hours at room temperature or overnight at 4 degrees C. 5 Wash and add fluorescent secondaryWash 3 times for 5 minutes. Add cross-adsorbed fluorescent secondary antibody at 1:500-1:1000 for 45-60 minutes protected from light. 6 Counterstain and mountWash 3 times. Add DAPI if desired. Mount with antifade medium and coverslip without bubbles. 7 Image and controlAcquire no-primary, single-color, and full-stain controls. Use identical image settings for samples that will be compared. Multiplex tissue stainingFor multiple primary antibodies, confirm species compatibility or use directly conjugated primaries or sequential staining.AutofluorescenceTissue autofluorescence can resemble true signal. Always compare to no-primary controls.
IHC optimization protocol Use this workflow when an antibody or tissue type needs condition screening before final staining. Use this format when: Signal is weak, background is high, staining is inconsistent, or the antibody is being validated in a new tissue.Optimize first: Positive tissue, retrieval pH, antibody dilution, incubation time, detection strength, and blocking strategy. 1 Choose control tissueUse a known positive tissue or cell pellet and a known negative tissue when available. Include the experimental tissue only after positive control staining is working. 2 Build a retrieval matrixTest no retrieval, citrate pH 6.0, and Tris-EDTA pH 9.0 when tissue availability allows. Use the same section thickness and slide type for all conditions. 3 Test antibody dilutionDilute 5-Methylcytosine (5-mC) Monoclonal Antibody [33D3] across a small matrix such as 1:50, 1:100, 1:250, and 1:500, or follow datasheet starting ranges. Keep retrieval and detection constant while testing dilution. 4 Compare incubation conditionsIf signal is weak, compare 1 hour at room temperature with overnight at 4 degrees C. Longer incubation can improve weak signal but may increase background. 5 Adjust detection strengthIf signal remains weak, increase polymer incubation within the detection system limits, use amplification, or extend chromogen development while monitoring background. 6 Reduce backgroundIncrease wash time, reduce antibody concentration, change blocking buffer, shorten chromogen development, or add detergent to wash buffer if compatible with the tissue and detection system. 7 Lock the protocolOnce the best condition is chosen, stain all comparative samples together using the same retrieval, antibody dilution, incubation time, detection system, and development time. One change at a timeChange one major variable at a time when troubleshooting so the cause of improvement is clear.DocumentationRecord lot numbers, retrieval buffer, heating method, antibody dilution, incubation time, detection system, and imaging settings.
Protein immunoprecipitation protocol Use this workflow to enrich a target protein from cell or tissue lysate using an antibody and protein A/G magnetic or agarose beads. Use this format when: Pulling down a target protein for Western blot, mass spectrometry, enzymatic assay, or interaction analysis.Optimize first: Lysis buffer stringency, lysate amount, antibody amount, bead amount, binding time, wash stringency, and elution method. 1 Prepare cold lysis bufferUse your own non-denaturing IP-compatible lysis buffer or a buffer selected from the Western blot protocol that matches the sample type and target. As a starting point, use 20-50 mM Tris or HEPES pH 7.4-7.6, 150 mM NaCl, 1 percent NP-40 or Triton X-100, 1 mM EDTA, and fresh protease inhibitors such as Protease Inhibitor Cocktail when compatible with the downstream assay. Add phosphatase inhibitors for phospho or PTM targets. 2 Lyse sampleUse 0.5-2 mg total protein per IP as a starting range. Lyse cells on ice with cold lysis buffer containing fresh protease inhibitors. Incubate 15-30 minutes on ice with periodic mixing. For tissue, homogenize gently in cold lysis buffer. Clarify lysate at 12,000-16,000 x g for 10-15 minutes at 4 degrees C. 3 Pre-clear lysateAdd 20-30 uL washed protein A/G beads per IP to lysate and rotate 30-60 minutes at 4 degrees C. Collect supernatant and discard pre-clear beads to reduce nonspecific binding. 4 Bind antibody to targetAdd 2-5 ug 5-Methylcytosine (5-mC) Monoclonal Antibody [33D3] per 0.5-1 mg lysate as a starting point, or follow the datasheet. Rotate 2-4 hours at 4 degrees C, or overnight for low-abundance targets. 5 Capture immune complexesAdd 20-40 uL washed protein A/G magnetic or agarose beads per IP. Rotate 1-2 hours at 4 degrees C. Use protein A, protein G, or A/G according to antibody species and isotype. 6 Wash beadsWash beads 3-5 times with 0.5-1 mL cold lysis buffer. For high background, add one higher-salt wash such as 300-500 mM NaCl if the interaction or epitope can tolerate it. Keep beads cold and minimize bead loss. 7 Elute targetFor Western blot, add 20-40 uL 1x or 2x SDS sample buffer and heat 70-95 degrees C for 5-10 minutes depending on target stability and bead type. For native elution, use low pH glycine or competing peptide if compatible. 8 Analyze eluateRun input, flow-through if needed, wash control, IgG control, beads-only control, and IP eluate. For WB detection, use a secondary antibody strategy that minimizes IgG heavy-chain and light-chain interference. Antibody compatibilityWB validation does not guarantee IP performance. IP requires antibody binding under native or partially native conditions.Heavy chain interferenceUse crosslinked antibody beads, light-chain-specific secondary antibody, native elution, or antibodies from different species when heavy chain overlaps the target size.
Co-IP protocol Use this workflow to preserve and test protein-protein interactions during immunoprecipitation. Use this format when: Pulling down a bait protein and detecting associated partner proteins.Optimize first: Mild lysis conditions, salt concentration, detergent choice, lysate freshness, wash stringency, and reciprocal IP controls. 1 Use mild lysis conditionsPrepare cold mild lysis buffer such as 20-50 mM Tris or HEPES, 100-150 mM NaCl, 0.2-0.5 percent NP-40 or digitonin, 1 mM EDTA, and fresh inhibitors. Avoid harsh detergents and high salt unless the interaction is strong. 2 Lyse gentlyUse fresh cells when possible. Lyse on ice for 15-30 minutes with gentle mixing. Clarify lysate at 10,000-14,000 x g for 10 minutes at 4 degrees C. Avoid sonication unless necessary because it can disrupt complexes. 3 Pre-clearPre-clear lysate with 20-30 uL beads for 30-60 minutes at 4 degrees C. Transfer the supernatant to a new tube. 4 Add bait antibodyAdd 2-5 ug 5-Methylcytosine (5-mC) Monoclonal Antibody [33D3] per IP if it is the bait antibody. Rotate 2-4 hours at 4 degrees C. For weak interactions, overnight incubation can help but may increase nonspecific binding. 5 Capture complexesAdd 20-40 uL protein A/G beads and rotate 1-2 hours at 4 degrees C. Keep all steps cold. 6 Wash gentlyWash 3-4 times with mild lysis buffer. If nonspecific bands are high, increase salt stepwise to 200-300 mM NaCl or reduce detergent exposure based on complex stability. 7 Elute and detectElute with SDS sample buffer for Western blot. Detect bait and suspected partner proteins on separate blots or after stripping when appropriate. 8 Confirm interactionInclude IgG control, beads-only control, input lysate, and reciprocal Co-IP when possible. Validate that the partner signal is enriched in the bait IP compared with controls. Complex preservationCo-IP success depends heavily on keeping the complex intact. Use fresh lysate, cold buffers, and mild washing first.Nuclease optionFor DNA- or RNA-mediated associations, add nuclease controls when needed to distinguish direct protein interaction from nucleic-acid bridged association.
Crosslinked IP or chromatin-associated target protocol Use this workflow as a general crosslinked IP starting point for chromatin-associated proteins. For full ChIP workflows, see the dedicated ChIP protocol. Use this format when: A chromatin-associated protein or complex needs crosslink-stabilized enrichment before downstream analysis.Optimize first: Crosslinking time, chromatin fragmentation, antibody amount, wash stringency, reversal conditions, and DNA/protein recovery method.Alternative workflow: If the goal is target-specific protein-DNA enrichment rather than conventional crosslinked IP or ChIP, consider EpiNext CUT&LUNCH Assay Kit or EpiNext CUT&RUN-Fast Kit. 1 Crosslink sampleTreat cells with 1 percent formaldehyde for 10 minutes at room temperature as a starting condition. Quench with 125 mM glycine for 5 minutes. Wash cells with cold PBS containing inhibitors. 2 Prepare nuclei or lysateCollect cells and prepare nuclei or chromatin-enriched lysate using a compatible buffer. Keep samples cold and include protease inhibitors. 3 Fragment chromatin or complexesSonicate or digest to produce the fragment size needed for the downstream assay. For ChIP-qPCR, 200-800 bp DNA fragments are often used. Confirm shearing before large experiments. 4 Clarify and pre-clearClarify lysate and pre-clear with blocked protein A/G or ChIP-compatible beads for 30-60 minutes at 4 degrees C. 5 Add antibodyAdd 2-10 ug 5-Methylcytosine (5-mC) Monoclonal Antibody [33D3] per IP depending on target abundance and chromatin amount. Rotate overnight at 4 degrees C for chromatin-associated targets. 6 Capture and washAdd blocked beads and rotate 1-2 hours. Wash sequentially with low-salt, high-salt, LiCl or detergent wash, and TE buffer when compatible with the downstream method. 7 Elute and reverse crosslinksElute complexes and reverse crosslinks, commonly at 65 degrees C for 2-6 hours or overnight depending on protocol. Treat with RNase and proteinase K if recovering DNA. 8 Purify and analyzePurify DNA or protein as required. Analyze by qPCR, sequencing library preparation, Western blot, or other downstream method. Dedicated ChIP pageFor optimized native and crosslinking ChIP details, see the dedicated ChIP protocol.CrosslinkingOver-crosslinking can reduce antibody access and recovery. Start with mild crosslinking and optimize.CUT&LUNCH or CUT&RUN-Fast optionFor protein-DNA interaction profiling from cells or tissues, use EpiNext CUT&LUNCH Assay Kit or EpiNext CUT&RUN-Fast Kit only when that assay format fits the experimental goal.
RNA immunoprecipitation protocol Use this workflow to enrich RNA-associated proteins or protein-bound RNA while preserving RNA integrity. Use this format when: Studying RNA-binding proteins, protein-RNA complexes, or RNA associated with a target protein.Optimize first: RNase-free handling, lysis conditions, antibody amount, salt concentration, wash stringency, and RNA purification.Kit option: For a complete RIP workflow with coordinated reagents, use EpiNext CUT&LUNCH RNA Immunoprecipitation (RIP) Kit when its sample type and downstream readout match the experiment. 1 Prepare RNase-free workspaceUse RNase-free tubes, filter tips, buffers, and gloves. Add RNase inhibitor to lysis, wash, and binding buffers when compatible. 2 Lyse cells gentlyLyse cells in RIP lysis buffer containing mild detergent, physiological salt, protease inhibitors, and RNase inhibitor. Use fresh lysate when possible. Clarify at 12,000-16,000 x g for 10 minutes at 4 degrees C. 3 Pre-clear lysatePre-clear lysate with washed beads for 30-60 minutes at 4 degrees C. Transfer supernatant to a new RNase-free tube. 4 Add antibodyAdd 2-10 ug 5-Methylcytosine (5-mC) Monoclonal Antibody [33D3] per RIP depending on target abundance. Rotate 2-4 hours or overnight at 4 degrees C. Include IgG control and input RNA controls. 5 Capture complexesAdd 20-40 uL protein A/G beads that have been washed and blocked with BSA, yeast tRNA, or other compatible blocking reagent. Rotate 1-2 hours at 4 degrees C. 6 Wash under RNase-free conditionsWash 4-6 times with cold RIP wash buffer. Increase salt only if nonspecific RNA background is high and the interaction can tolerate it. 7 Recover RNADigest protein or reverse crosslinks if used, then purify RNA using phenol/chloroform, column purification, or magnetic bead cleanup. Include DNase treatment when downstream qPCR could detect DNA contamination. 8 Analyze RNAAnalyze enriched RNA by RT-qPCR, sequencing, or other downstream assay. Normalize to input and compare to IgG control. RNase controlRNA loss is often caused by RNase contamination. Use RNase inhibitor and work quickly on ice.Input controlSave 1-10 percent input before IP for normalization.RIP kit optionFor a kit-based RNA immunoprecipitation workflow, consider EpiNext CUT&LUNCH RNA Immunoprecipitation (RIP) Kit when a complete RIP reagent set is preferred.
Cultured cell IF/ICC protocol Use this workflow for fluorescent antibody staining of adherent or suspension cells on coverslips, chamber slides, or imaging plates. Use this format when: Detecting protein localization, nuclear markers, cytoplasmic proteins, histone marks, signaling proteins, or cell morphology in cultured cells.Optimize first: Fixation method, permeabilization strength, blocking buffer, primary antibody dilution, and imaging exposure. 1 Plate cellsSeed cells onto sterile coverslips, chamber slides, or imaging plates so they reach 50-80 percent confluence on staining day. For coverslips in a 24-well plate, use enough medium to fully cover the cells. 2 Fix cellsRemove medium and gently rinse once with PBS. Add 4 percent paraformaldehyde in PBS for 10-15 minutes at room temperature. For some cytoskeletal, membrane, or epitope-sensitive targets, compare with cold methanol fixation for 5-10 minutes at -20 degrees C. 3 Wash and permeabilizeWash 3 times with PBS, 5 minutes each. Permeabilize with 0.1-0.3 percent Triton X-100 in PBS for 5-10 minutes at room temperature. For nuclear histone or chromatin targets, 0.2-0.5 percent Triton X-100 can be tested. For membrane targets, reduce or omit detergent. 4 Block nonspecific bindingAdd blocking buffer, such as 1-5 percent BSA or 5 percent normal serum in PBS with 0.05-0.1 percent Tween-20. Incubate 30-60 minutes at room temperature. Use serum from the host species of the secondary antibody when practical. 5 Add primary antibodyDilute 5-Methylcytosine (5-mC) Monoclonal Antibody [33D3] in blocking buffer. Start with the datasheet-recommended IF/ICC dilution or test 1:100, 1:250, and 1:500. Add enough volume to cover the sample, such as 100-200 uL per coverslip or 50-100 uL per chamber well. Incubate 1 hour at room temperature or overnight at 4 degrees C in a humidified chamber. 6 Wash after primary antibodyWash 3 times with PBS or PBST, 5 minutes each, using gentle rocking. Do not allow cells to dry. 7 Add fluorescent secondary antibodyDilute fluorophore-conjugated secondary antibody in blocking buffer, commonly 1:500-1:1000. Add enough volume to cover the cells and incubate 45-60 minutes at room temperature protected from light. 8 Counterstain and mountWash 3 times. Add DAPI or another nuclear counterstain for 5 minutes if desired, then wash once. Mount coverslips with antifade mounting medium and cure according to the mountant instructions. 9 Image and document settingsAcquire images using the same exposure, laser power, gain, and offset for samples that will be compared. Include no-primary and secondary-only controls to evaluate background. Fixation choiceParaformaldehyde preserves morphology well. Methanol can improve some cytoskeletal or nuclear epitopes but may disrupt membrane morphology.AutofluorescenceIf background is high, reduce fixation time, improve washing, test a different blocking buffer, or switch fluorophores.
Tissue immunofluorescence protocol Use this workflow for fluorescent antibody staining of frozen, fixed frozen, or FFPE tissue sections. Use this format when: Detecting spatial protein expression in tissue sections with fluorescence rather than chromogenic IHC.Optimize first: Section type, antigen retrieval, autofluorescence control, permeabilization, antibody dilution, and mounting medium. 1 Prepare sectionsUse 4-8 um tissue sections mounted on charged slides. Air dry frozen sections for 10-20 minutes. For FFPE sections, deparaffinize through xylene and graded ethanol, then rehydrate to water. 2 Fix or retrieve antigenFor frozen sections, fix with cold acetone for 10 minutes or 4 percent paraformaldehyde for 10-15 minutes depending on the target. For FFPE sections, perform heat-induced antigen retrieval in citrate buffer pH 6.0 or Tris-EDTA pH 9.0 for 10-20 minutes, then cool 20 minutes. 3 Reduce autofluorescence if neededFor tissue with high autofluorescence, treat with an autofluorescence quenching reagent or freshly prepared 0.1 percent sodium borohydride for 5-10 minutes, followed by thorough PBS washes. Choose the method compatible with your fluorophores. 4 Permeabilize and blockPermeabilize with 0.1-0.3 percent Triton X-100 in PBS for 5-10 minutes if intracellular or nuclear access is needed. Block 30-60 minutes with 5 percent normal serum or 1-5 percent BSA in PBS. 5 Add primary antibodyDilute 5-Methylcytosine (5-mC) Monoclonal Antibody [33D3] in blocking buffer. Start with the datasheet-recommended tissue IF dilution or test 1:100, 1:250, and 1:500. Apply 100-200 uL per section, cover with parafilm or use a humidified chamber, and incubate 1-2 hours at room temperature or overnight at 4 degrees C. 6 Wash and add secondary antibodyWash 3 times for 5 minutes each in PBS or PBST. Add fluorophore-conjugated secondary antibody diluted 1:500-1:1000. Incubate 45-60 minutes at room temperature protected from light. 7 Counterstain and mountWash 3 times. Add DAPI if desired. Mount with antifade medium and coverslip carefully to avoid bubbles. 8 Image sectionsUse identical microscope settings for comparative samples. Include no-primary controls and known positive tissue when available. Tissue handlingDo not let sections dry after rehydration or blocking begins.RetrievalSome epitopes require retrieval for FFPE tissue but not frozen tissue. Test pH 6 and pH 9 retrieval when starting a new antibody.
Multiplex IF/ICC protocol Use this workflow to detect two or more targets in the same cells or tissue section using spectrally distinct antibodies. Use this format when: Comparing colocalization, marker identity, signaling state, or target expression in the same sample.Optimize first: Host species compatibility, fluorophore separation, antibody order, blocking, cross-reactivity controls, and exposure settings. 1 Plan antibody panelChoose primary antibodies from different host species when possible. Match secondary antibodies to non-overlapping fluorophores. Avoid fluorophores with heavy spectral overlap unless spectral unmixing is available. 2 Fix, permeabilize, and blockPrepare the sample using the IF/ICC or tissue IF fixation and blocking conditions appropriate for the most sensitive target. Use enough blocking buffer to reduce nonspecific secondary antibody binding. 3 Add primary antibody mixPrepare a master mix containing each primary antibody at its optimized dilution. Include 5-Methylcytosine (5-mC) Monoclonal Antibody [33D3] if it is one of the targets in the panel. Start with single-stain optimized dilutions before combining antibodies. Incubate 1 hour at room temperature or overnight at 4 degrees C. 4 Wash thoroughlyWash 3 times for 5 minutes each with PBS or PBST. Extend washes to 5-10 minutes when background or cross-channel signal is high. 5 Add secondary antibody mixAdd cross-adsorbed secondary antibodies diluted in blocking buffer, commonly 1:500-1:1000. Incubate 45-60 minutes protected from light. 6 Counterstain and mountWash 3 times. Add nuclear counterstain if needed, then mount with antifade medium compatible with all fluorophores. 7 Acquire controlsImage single-color controls, no-primary controls, and full-stain samples using consistent settings. Use single-color controls to identify bleed-through or compensation needs. Same-species primariesIf two primary antibodies share the same host species, use directly conjugated primaries, sequential staining with blocking, Fab fragments, or tyramide signal amplification when appropriate.ColocalizationDo not call colocalization from overexposed images. Keep all channels below saturation.
Phospho/PTM IF protocol Use this workflow for phosphorylation, acetylation, methylation, ubiquitination, or other modification-specific antibody staining. Use this format when: Detecting transient signaling or post-translational modifications in cells or tissue.Optimize first: Sample timing, inhibitor use, fixation speed, permeabilization, antibody dilution, and positive or negative treatment controls. 1 Prepare treated and control samplesPlan untreated, stimulated, inhibited, knockdown, or positive control samples before staining. For phospho targets, add phosphatase inhibitors during washes or lysis-related steps where applicable and fix quickly after treatment. 2 Fix rapidlyRemove medium and rinse briefly with warm PBS when needed. Fix with 4 percent paraformaldehyde for 10-15 minutes at room temperature, or use methanol fixation for targets known to work better with alcohol fixation. 3 Permeabilize for nuclear or intracellular accessWash 3 times. Use 0.1-0.3 percent Triton X-100 for 5-10 minutes. For histone or chromatin PTMs, test 0.2-0.5 percent Triton X-100 and consider a short pre-extraction step only if compatible with morphology and the target. 4 BlockBlock 45-60 minutes with 1-5 percent BSA in PBS or TBS. For phospho-specific antibodies, BSA is often preferred over milk because milk can contain phosphoproteins that increase background. 5 Add primary antibodyDilute 5-Methylcytosine (5-mC) Monoclonal Antibody [33D3] in blocking buffer. Start with the datasheet-recommended IF dilution or test 1:100, 1:250, and 1:500. Incubate overnight at 4 degrees C for low-abundance PTMs or weak signals. 6 Wash and detectWash 3 times for 5 minutes. Add fluorophore-conjugated secondary antibody at 1:500-1:1000 for 45-60 minutes protected from light. Wash 3 times. 7 Mount and imageCounterstain if needed, mount with antifade medium, and image positive and negative controls with the same settings. For signal quantification, avoid saturated pixels and analyze the same cellular compartment across samples. PTM specificityUse inhibitor, stimulation, peptide competition, knockdown, or known positive/negative controls when available to support modification-specific interpretation.Blocking bufferIf background is high, compare BSA, serum, fish gelatin, or commercial antibody diluent.
Sandwich ELISA protocol Use this workflow when a target is captured by one antibody and detected by a second antibody that recognizes a different epitope. Use this format when: Quantifying low-abundance soluble antigens in complex biological matrices where a compatible matched antibody pair is available.Optimize first: Capture antibody concentration, detection antibody concentration, sample dilution, blocking buffer, wash stringency, standard curve range, and matrix effects. Sample typesBest sample types: Serum, plasma, saliva, culture supernatants, and tissue homogenates.Best for: Quantifying low-abundance antigens, such as cytokines, hormones, and biomarkers, in complex biological matrices.Accuracy note: Sandwich ELISA is usually not the best choice for very small or single-epitope analytes because two non-overlapping antibody binding sites are needed. 1 Prepare plate map and reagentsPlan standards, blanks, negative controls, positive controls, and samples before starting. Bring coated plate, standards, samples, wash buffer, blocking buffer, detection antibody, enzyme conjugate, substrate, and stop solution to room temperature unless the kit manual says otherwise. For manual coating, use a high-binding 96-well ELISA plate. 2 Coat capture antibodyDilute capture antibody in carbonate-bicarbonate coating buffer, pH 9.6, or the buffer specified for the antibody pair. Add 100 uL per well. Cover the plate and incubate overnight at 4 degrees C, or 2 hours at room temperature as a faster starting condition. Aspirate the coating solution after incubation. 3 Wash and blockWash 3 times with 250-300 uL per well 1x PBST or TBST. Add 200 uL per well blocking buffer, such as 1-5 percent BSA, casein, or nonfat dry milk in PBS/TBS. Incubate 1 hour at room temperature. Wash 3 times. 4 Add standards and samplesPrepare standards in duplicate or triplicate using the recommended diluent. Dilute samples so readings fall within the linear range. For cell lysates or tissue homogenates, prepare clarified samples and include fresh protease inhibitor during upstream extraction when compatible with the assay, such as R-1101 Protease Inhibitor Cocktail. Add 100 uL per well and incubate 1-2 hours at room temperature, or overnight at 4 degrees C for low-abundance targets. Wash 4-5 times. 5 Add detection antibodyDilute detection antibody in assay diluent. If this product is used as the detection antibody, add 5-Methylcytosine (5-mC) Monoclonal Antibody [33D3] at the datasheet-recommended dilution or start at 0.25-2 ug/mL for optimization. Add 100 uL per well and incubate 1 hour at room temperature. Wash 4-5 times. 6 Add enzyme conjugate and substrateAdd 100 uL per well HRP-conjugated secondary antibody, or R-1098 Streptavidin: HRP Conjugate when the detection antibody or binding reagent is biotin-labeled. Incubate 30-60 minutes at room temperature protected from light when needed. Wash 5 times, then add 100 uL TMB substrate per well. 7 Stop and readDevelop color for 5-20 minutes until the standard curve is visible but not saturated. Add 50-100 uL stop solution per well and read absorbance at 450 nm, with 570 or 620 nm correction if available. Analyze standards with a 4-parameter logistic curve when appropriate. ControlsInclude blank wells, zero standard, standard curve, matrix control, no-sample control, and positive sample when available.Wash consistencyMost high background in sandwich ELISA comes from insufficient washing, over-concentrated detection antibody, or incompatible blocking buffer.
Direct ELISA protocol Use this workflow when the target is immobilized or captured and detected directly by a labeled antibody, direct detection reagent, or kit-specific detection system. Use this format when: A direct detection kit, directly labeled antibody, purified antigen, semi-purified antigen, or target-specific direct detection reagent is available.Optimize first: Coating or sample input, target purity, detection reagent amount, incubation time, wash stringency, and substrate development time. Sample typesBest sample types: Purified or semi-purified antigens, recombinant proteins, and clarified cell or tissue lysates when target coating and background are acceptable.Best for: Screening antibody specificity or confirming the presence of an antigen in an isolated, high-purity state.Accuracy note: Direct ELISA is simple and fast, but it is less ideal for crude complex matrices because non-target proteins can compete for plate binding and increase background. 1 Prepare plate and samplesPlan standards, blanks, negative controls, positive controls, and sample dilutions. For manual direct ELISA, use purified or semi-purified antigen, recombinant protein, or clarified lysate only when plate coating and background are acceptable. Add 100 uL per well of standards or samples according to the kit format or coating requirement. 2 Immobilize target if requiredFor coated-plate direct ELISA, incubate antigen or sample in the well overnight at 4 degrees C or 2 hours at room temperature. For direct target-detection kits, follow the kit manual for binding, capture, or sample incubation conditions. 3 Wash and blockWash 3 times with 250-300 uL per well wash buffer. Add 200 uL per well blocking buffer for 30-60 minutes if the assay format requires blocking. Some direct detection kits include a proprietary blocking or assay buffer that should be used instead. 4 Add direct detection reagentAdd 100 uL per well of the directly labeled antibody, direct detection reagent, or enzyme-conjugated target-specific reagent. If using a directly conjugated antibody, start with the datasheet-recommended dilution or test 0.1-1 ug/mL. Incubate 30-90 minutes at room temperature protected from light when appropriate. 5 Wash and developWash 4-5 times to reduce background. Add 100 uL per well substrate. Develop until standards or positive controls show clear separation without saturation. 6 Stop and readAdd 50-100 uL stop solution when using TMB and read at 450 nm. Use the correction wavelength recommended for the plate reader if available. Compare results to the standard curve or kit-specific calculation method. Direct detection kitsSome EpigenTek assay kits use direct target-detection formats for small molecules, DNA/RNA damage markers, or epigenetic modifications. In those cases, the kit manual overrides generic antibody-only workflow assumptions.Background controlInclude blank, no-target, no-detection-reagent, and positive control wells when compatible with the kit or assay design.
Indirect ELISA protocol Use this workflow to detect antibody binding to an immobilized antigen or target molecule using an unconjugated primary antibody and labeled secondary antibody. Use this format when: Measuring antibody binding, total antibody concentration, titer, or immune response to a coated antigen.Optimize first: Antigen coating concentration, sample or antibody dilution, blocking buffer, secondary antibody dilution, and wash stringency. Sample typesBest sample types: Serum, plasma, saliva, and other body fluids.Best for: Measuring total antibody concentrations, titers, or screening immune responses against specific pathogens or antigens.Accuracy note: Match the secondary antibody to the sample antibody species and isotype, and validate saliva or other non-serum matrices before relying on quantitative comparisons. 1 Coat antigen or targetDilute purified antigen, peptide, protein, histone, modified target, cell lysate, or other coating material in carbonate-bicarbonate buffer or PBS. Add 100 uL per well. A common starting range is 0.5-5 ug/mL for purified protein or peptide. If the antigen or capture reagent is biotinylated, a streptavidin-coated plate such as R-1102 Streptavidin-Coated Strip Microwell Plate may be used instead of passive coating. Incubate overnight at 4 degrees C or 2 hours at room temperature. 2 Wash and blockAspirate coating solution and wash 3 times with 250-300 uL per well PBST or TBST. Add 200 uL per well blocking buffer. Incubate 1 hour at room temperature, then wash 3 times. 3 Add primary antibodyDilute 5-Methylcytosine (5-mC) Monoclonal Antibody [33D3] in assay diluent. Start with the datasheet-recommended dilution when available; otherwise test a dilution series such as 1:250, 1:500, 1:1000, and 1:2000. Add 100 uL per well and incubate 1 hour at room temperature or overnight at 4 degrees C for weak targets. 4 Wash after primary antibodyWash 4-5 times with 250-300 uL per well wash buffer. Tap the inverted plate on clean absorbent material after the final wash to remove residual liquid without drying the wells. 5 Add enzyme-conjugated secondary antibodyDilute HRP- or AP-conjugated secondary antibody in assay diluent, commonly 1:2000-1:10000 depending on supplier and signal strength. For common mouse or rabbit primary antibodies, host-matched options include A12003 HRP-Goat Anti-Mouse Secondary Antibody or A12004 HRP-Goat Anti-Rabbit Secondary Antibody. Add 100 uL per well and incubate 30-60 minutes at room temperature. 6 Develop signalWash 5 times. Add 100 uL substrate per well. For HRP/TMB, develop 5-20 minutes protected from strong light, then stop with 50-100 uL stop solution and read at 450 nm. 7 Interpret resultsSubtract blank background and compare signal across antigen-coated, uncoated, no-primary, and no-secondary controls. High signal in no-primary wells suggests secondary antibody or blocking-related background. Antigen coatingToo much coated antigen can increase background. If the signal is high but nonspecific, reduce coating concentration before changing everything else.Primary antibody variableThe product page can replace the primary antibody variable in this protocol with the selected antibody name.
Competitive ELISA protocol Use this workflow when sample target competes with immobilized or labeled target for antibody or binding reagent. Use this format when: The analyte is small, has one dominant epitope, or is measured by competition rather than two-antibody sandwich capture.Optimize first: Sample dilution, competitor concentration, antibody concentration, incubation time, standard curve range, and matrix interference. Sample typesBest sample types: Serum, plasma, saliva, culture supernatants, and crude biological fluids.Best for: Detecting small molecules, hormones, or targets with only a single antigenic epitope.Accuracy note: Competitive ELISA produces an inverse signal, so higher analyte levels usually give lower absorbance or fluorescence. 1 Prepare standards and samplesPrepare a full standard curve using the assay diluent recommended for the target. For serum, plasma, saliva, culture supernatants, or crude biological fluids, dilute samples into the same matrix when possible. Competitive assays often need careful sample dilution because high target concentration produces lower signal. 2 Add standards, samples, and competitorAdd 50-100 uL per well of standards or samples according to the assay design. Add competitor, tracer, or labeled target reagent if the protocol requires a pre-mix. Mix gently without splashing. 3 Add antibody or binding reagentAdd the antibody or target-specific binding reagent at the optimized concentration. When using an antibody-based format, add 5-Methylcytosine (5-mC) Monoclonal Antibody [33D3] only if the product is the intended competitive binding antibody and follow the datasheet dilution first. 4 Incubate competition reactionIncubate 1-2 hours at room temperature, or as specified by the kit manual. Keep incubation times consistent across the plate because competitive assays are especially sensitive to timing differences. 5 Wash thoroughlyWash 4-5 times with 250-300 uL per well wash buffer. Avoid drying the wells. Inconsistent washing can distort the inverse standard curve. 6 Develop and readAdd 100 uL substrate per well and develop until the low-analyte standards produce strong but unsaturated signal. Stop and read at the appropriate wavelength. Fit the data using the curve model recommended by the kit or assay format. Signal directionIn competitive ELISA, stronger sample target usually means lower signal. Label graphs and reports clearly to avoid reversed interpretation.TimingUse a multichannel pipette and consistent timing between rows when possible.





 Cart (0)
Cart (0)


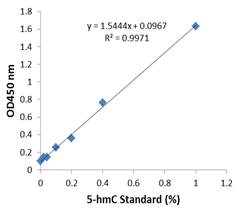
![5-Hydroxymethylcytosine (5-hmC) Monoclonal Antibody [HMC/4D9]](images/a1018data.gif)