 Cart (0)
Cart (0)


In the early 1960s, Vincent G. Allfrey and his colleagues at the Rockefeller Institute (today’s Rockefeller University) demonstrated by way of C14 labeling the incorporation of methyl and acetyl groups into histones.1 The evidence suggested that these chemical additions succeeded protein synthesis, although the means by which they were incorporated as well as the extent of their biological significance were not fully understood at the time. Allfrey’s pioneering work effectively laid the foundation for histone post-translational modification (PTM) research, with future studies exposing additional histone marks, their modifying enzymes, and the crucial roles they serve in controlling gene expression.
Making their mark
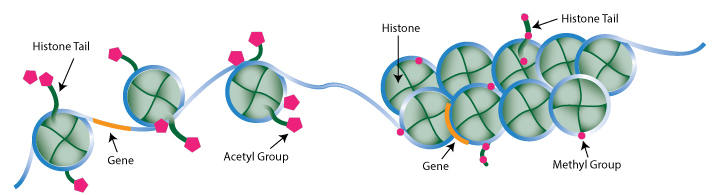
Once regarded as nothing more than mere packaging units that condensed DNA into chromosomes, histones have been revealed to support critical cellular processes(transcription, DNA replication, and DNA repair, to name a few) via diverse PTMs that regulate their interactions with DNA and other nuclear proteins. Histone marks come in a variety of chemistries, collectively constituting a so-called “histone code” involved in the epigenetic regulation of chromatin. Covalent modifications to histone tails, including methylation, acetylation, ubiquitination, sumoylation, citrullination, and phosphorylation, are well-correlated with chromatin structure and accessibility, which in turn impact transcriptional activation/silencing.

Abnormal PTM patterns are linked to various pathologies such as cancer, autoimmune disorders, and inflammatory and neurological diseases. As Dr. Arif Hussain, Professor and Chairperson of the School of Life Sciences at Manipal Academy of Higher Education in Dubai, points out, “Yes, a great deal of research is directed towards histone modification and chromatin accessibility, particularly in different diseases (cancer, diabetes, etc.). Histone modifications are also being explored as potential biomarkers of disease progression and prognosis, most so for cancer. However, out of the enormous list of histone modification marks, only a few have been studied, which requires a great deal of attention.”Thus, reliable methods to detect these modifications would provide useful information concerning histone PTM-associated disease processes and histone PTM-targeted drug development.
Dr. Catherine Musselman, Associate Professor in the Department of Biochemistry and Molecular Genetics and Director of the Structural Biology and Biochemistry Program at the University of Colorado Anschutz Medical Campus, notes, “There was certainly a big shift [in epigenetic research] around 1999-2000 wherein a huge effort was mounted to start studying histone modifications and their role in genome regulation, especially surrounding the idea of the so-called histone code. And definitely as it became clear that these pathways are druggable a huge effort was placed into the development of related therapeutics.”
Deciphering the code
When it comes to assessing histone PTMs, a multitude of analytical tools and techniques are at an investigator’s disposal. In general, these methods can be categorized as either immunoassays or antibody-free assays. Classical antibody-based assays like western blotting, immunohistochemistry, immunocytochemistry, immunofluorescence, and enzyme-linked immunosorbent assays (ELISAs) have long-standing and widespread utility in proteomics, especially for the detection of histone PTMs. ELISA, in particular, offers the added benefit of multiplexing capabilities, accommodating the simultaneous quantification of assorted modifications on a single microplate. In addition to histone PTM quantification, ELISAs are also employed to quantify enzyme activity levels of histone PTM writers and erasers such as histone acetyltransferases and deacetylases, both validated candidates for drug treatment of cancer and other diseases.
For studying DNA-histone interactions, chromatin immunoprecipitation, or ChIP, is the popular go-to approach. By coupling ChIP with varied downstream applications such as PCR (ChIP-PCR), DNA microarray (ChIP-chip), and next generation sequencing(ChIP-seq), one has the luxury of tailoring their experimental output to identify, on either a region-specific or genome-wide level, the DNA binding sites for histones containing modifications of interest.
Recent advances in ChIP technology, namely cleavage under targets and release using nuclease (CUT&RUN)3 and cleavage under targets and tagmentation (CUT&Tag)4, afford several improvements over traditional chromatin immunoprecipitation in terms of starting input amounts and mapping resolution.
According to Dr. Peter Tessarz, Group Leader of the Chromatin and Ageing research group at the Max Planck Institute for Biology of Ageing in Cologne, Germany, “Methods like CUT&RUN and CUT&Tag have gained a lot of attention, particularly with the possibility to move towards rare cell populations and/or even single cells. This obviously enables genome-wide mapping in precious samples that was impossible before.”
Radiolabeling (not unlike Allfrey and coworkers’ usage of acetate-2-C14 and methionine-methyl-C14 to track histone acetylation and methylation, respectively) and mass spectrometry are among the more notable antibody-free histone PTM assessment methods. Mass spectrometry appears the preferred choice for identifying and quantifying relative histone PTM abundance on a large scale.2 And by integrating high-resolution analyzers, mass spectrometry has the capability of discerning different histone PTMs with similar masses, making it possible to quantify both single and combinatorial PTM codes.
“It is challenging to correctly describe the landscape of histone PTMs, and in particular to confidently assign modifications to specific amino acids,”states Dr. Hussain. “However, mass spectrometry-based analyses of histone proteins can be used in detecting and quantifying histone variants and modifications which can be supplemented with computational biology/bioinformatics.”
In Dr. Musselman’s case, “We primarily use NMR spectroscopy to study histone modification readout in the context of the nucleosome. NMR is very powerful for looking at the conformational dynamics of the nucleosome and binding of various factors.”Indeed, nuclear magnetic resonance (NMR) studies have been elucidating the structure and interactions of histones since the 1970s, providing important data in support of the histone code hypothesis.5 “There has been a huge effort by many groups recently to better understand things mechanistically in the context of the nucleosome, which has always been challenging. Most notably, advances in NMR spectroscopy and high resolution single particle cryo-EM have allowed for some very exciting studies of the nucleosome and nucleosome interactions.
“NMR is very powerful for understanding how the conformational dynamics of the nucleosome are regulated by histone modifications. In addition, there have been a number of structures solved of remodelers and modifiers in complex with nucleosomes. This is extremely exciting as it is allowing us to gain a lot of insight into the mechanism of function of these complexes, as well as to better understand how histone modifications may regulate their activity.”
Modifications still needed
Of course, the aforementioned repertoire of methods is not without its drawbacks, leaving areas for improvement. Decoupling of extracted histones from their original DNA location in mass spectrometry protocols precludes defining the genome-wide distribution of histone marks. Hitches regarding specificity and cross-reactivity are unremitting concerns when utilizing antibodies. “Reliability of antibodies is always an issue and batch-testing would be fantastic,”remarks Dr. Tessarz.
ChIP-seq requires a large amount of input material to produce a strong enough signal over background noise, while the more advanced ChIP-related methods like CUT&RUN and CUT&Tag are both offered at a high cost. CUT&RUN necessitates expensive PA/MNase fusion protein that has significant A/T sequence bias, causing the target protein-interacted DNA region profiles to be seriously affected by the level of MNase digestion. CUT&Tag displays the same digestion bias and is less specific due to off-target accessibility of chromatin caused by the Tn5 transposase enzyme.
“We routinely use ChIP-seq and CUT&RUN,”Dr. Tessarz continues,“depending on cell number, antibody, and the need for cross-linking.Each of these methods has its limitation (cell number, harshness of washing, coverage), but this typically relies on a number of variables and is specific for each experiment.”
As for Dr. Musselman,“[NMR] definitely does have its limitations. Mainly, that it can be difficult to interpret this in terms of exact structural models. We all need to move towards using the full nucleosome in our studies. However, making modified nucleosomes in large quantities is very challenging both technically and in terms of cost. Improving technology for generating large quantities of modified nucleosomes that are cost appropriate for the typical research laboratory would do a lot to advance our understanding of histone PTMs.”
Despite their shortcomings, these methods have surely been indispensable in enhancing our knowledge of histone PTMs and their key functionalities in human physiology and disease. Consequently, histone deacetylase (HDAC) inhibitors at present constitute the bulk of the epigenetic drug market. Antibody-based strategies like ELISA offer a quick, convenient, and economical means to screen inhibitors of HDACs and other histone modifying enzymes in a high throughput manner, although a combination of different techniques is essential for a more comprehensive assessment of the effects of such drug compounds. Still, Further refinements to the current methodologies and future innovations in the way histone PTMs are surveyed will undoubtedly facilitate advancements in therapeutic treatments targeting these modifications.
References
- ALLFREY VG, FAULKNER R, MIRSKY AE. (1964). ACETYLATION AND METHYLATION OF HISTONES AND THEIR POSSIBLE ROLE IN THE REGULATION OF RNA SYNTHESIS. Proc Natl Acad Sci U S A. 1964; 51(5).786-794.
- Stransky S, Aguilan J, Lachowicz J, Madrid-Aliste C, Nieves E, Sidoli S. (2020). Mass Spectrometry to Study Chromatin Compaction. Biology (Basel). 2020;9(6):140.
- Skene PJ, Henikoff S. (2017) An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife.
- Kaya-Okur HS, Wu SJ, Codomo CA, et al. (2019). CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat Commun. 2019; 10(1):1930.
- van Emmerik CL, van Ingen H. (2019) Unspinning chromatin: Revealing the dynamic nucleosome landscapeby NMR. Prog Nucl Magn Reson Spectrosc. 110, 1-19.