 Cart (0)
Cart (0)


Unraveling the histone code is of utmost importance to many epigenetic researchers as they begin to understand how the combination of histone amino-terminal modifications forms a pattern, or code, that influences gene expression and many chromatin-templated processes. Investigating this complex epigenetic marking system can reveal the far-reaching effects of histone modifications on cell fate and disease development. Through a process called chromatin remodeling, a protein-DNA complex is loosened or tightened, thereby opening or closing certain genes to transcription.
Chromatin Remodeling
Chromatin is a protein-DNA complex found in eukaryotes that contains all the genetic information of the organism. It is composed of DNA tightly wrapped around a histone octamer, or two copies of four histone proteins H2A, H2B, H3 and H4. This DNA-histone complex is called the nucleosome, and it allows all genetic information to fit into the nucleus of every eukaryotic cell. In order for the cell to express certain genes, the chromatin must be made accessible to transcription factors. Chromatin can be modified by epigenetic mechanisms like histone modifications during transcription in order to inhibit or express certain genes. This process is called chromatin remodeling.
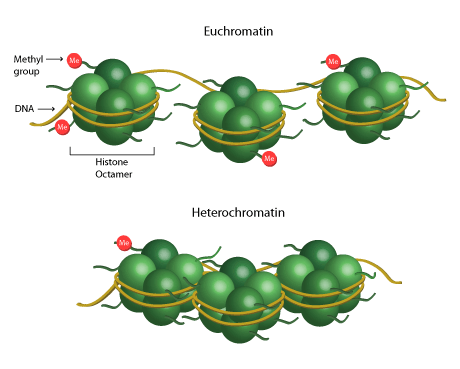
Chromatin remodeling is a biological process that plays a leading role in gene expression, DNA repair, and apoptosis. The DNA is negatively charged due to the numerous negatively charged phosphates in the backbone. Conversely, histone proteins are positively charged, so the DNA and histone proteins are naturally able to wrap together.
It is hypothesized that the remodeling of chromatin is determined by the histone code, which is a heavily complex theory indicating that the combination of post-translational histone modifications directly affect genetic transcription and expression. The hypothesis implies that there is a blueprint made by specific enzymes that allow the addition or removal of methyl or acetyl groups to histones, or to identify domains on the histone to be epigenetically altered, ultimately altering gene expression.
The histone code is incredibly complex, considering there are 19 known lysines on histone H3 alone known to be methylated, and each can be unmethylated, monomethylated, dimethylated, or trimethylated. There are numerous other modifications including lysine acetylation, arginine methylation, threonine/serine/tyrosine phosphorylation on histone H3. Additional modifications occurring to other histone proteins also must be considered in the complexity of the histone code.
Epigenetic Enzymes
The histone modification enzymes involved in the histone code include HATs (histone acetyltransferase), HMTs (histone methyltransferase) and HDACs (histone deacetylase). These enzymes are able to add or remove epigenetic marks to the DNA in order to rearrange the chromatin from a less active condensed state (heterochromatin) to a more active looser state (euchromatin), or vice versa. This changes the accessibility of the chromatin to transcription factors or DNA binding proteins during transcription, resulting in the silencing or expression of genes.
HATs add an acetyl group to a histone lysine, which allows the nucleosome to unravel enough to ready the DNA for transcription. Acetyl groups are negative, so this will make their grip on DNA much looser. Similarly, histone phosphorylation will relax the chromatin structure due to the negative charge of the phosphate group.
HMTs add one, two, or three methyl groups to either a lysine or an arginine on a histone tail, which typically leads to gene silencing. The addition of methyl groups will normally make the negatively charged histones more hydrophobic, which leads the molecules to stick together, thereby tightening the grip on DNA.
HDACs are responsible for removing the acetyl groups, which reverses the work done by the HAT to loosen the chromatin. It is crucial for the histone modification process to be heavily regulated, because an imbalance in the acetylation/methylation equilibrium could lead to cancer development and progression of diseases.
See a PDF list of popular histone modifications, including histone methylation, acetylation, phosphorylation, ubiquitination, and SUMOylation.
 For quantitative chromatin analysis and determination of euchromatin or
heterochromatin states use the EpiQuik Chromatin
Accessibility Assay Kit. This kit is ideal for gene-specific analysis of chromatin accessibility including
nucleosome/transcription factor positioning from various biological samples via real time PCR.
For quantitative chromatin analysis and determination of euchromatin or
heterochromatin states use the EpiQuik Chromatin
Accessibility Assay Kit. This kit is ideal for gene-specific analysis of chromatin accessibility including
nucleosome/transcription factor positioning from various biological samples via real time PCR.
Chromatin Remodeling and Pathological Development
Chromatin remodeling and histone modifications play a large part in disease development, particularly breast cancer development. Researchers at Cold Spring Harbor Labs identified the gene BPTF to be a chromatin-regulating protein present in the regulatory system of stem cells—particularly mammary stems cells. This gene is linked to the opening up of chromatin allowing for gene expression, which is crucial to the stem cell renewal process of bypassing the normal “stop signals” that mark the cell for death.
The researchers discovered that if the BPTF gene is turned off or removed, the stem cells could no longer renew themselves, and were permanently differentiated, meaning that they would also carry out the normal cell death process.1 This discovery could potentially be applied to cancer treatment since cancer cells share similarities in the ability to neglect the reproduction stop signs.
Understanding what leads to chromatin remodeling and what the histone patterns accomplish as part of the overall biological system and regulation of gene expression brings researchers closer to understanding the histone code. The implications for human biology and disease, including aging and cancer, are far-reaching.
References:
- Frey, Wesley D. et al., BPTF Maintains Chromatin Accessibility and the Self-Renewal Capacity of Mammary Gland Stem Cells. Stem Cell Reports, 9(1), 23-31.