Enzymatic targeted cleavage methods such as CUT&RUN and CUT&Tag have transformed epigenomics research. Their low cell input requirements, streamlined workflows, elimination of sonication, and compatibility with many histone modification and transcription factor targets have made them attractive alternatives to traditional ChIP-seq. For many applications, these methods provide faster sample processing and strong sensitivity from limited biological material.
At the same time, a growing body of benchmark studies, negative-control analyses, and computational correction efforts has highlighted an important technical consideration: CUT&RUN and CUT&Tag datasets can contain accessibility-associated background signal. This does not mean these methods are unsuitable. Rather, it means researchers should understand where open chromatin-associated signal can complicate interpretation, especially when studying targets with weak enrichment, broad distribution, or biologically meaningful occupancy outside highly accessible regulatory regions.
The source of accessibility-associated background differs between CUT&RUN and CUT&Tag. In CUT&RUN, the original method was designed to minimize background by performing brief, low-temperature MNase cleavage before the tethered nuclease could diffuse and cleave accessible chromatin. Later negative-control studies, however, show that recurrent high-signal regions can still appear in CUT&RUN datasets and may persist after standard peak calling and ENCODE blacklist filtering. In CUT&Tag, the concern is more direct because Tn5 has inherent affinity for exposed DNA, and elevated salt conditions are commonly used to reduce non-specific pA-Tn5 or pAG-Tn5 binding to accessible chromatin regions. (CUT&RUN, eLife 2017; CUT&Tag, Nature Communications 2019; CUTAC, eLife 2020)
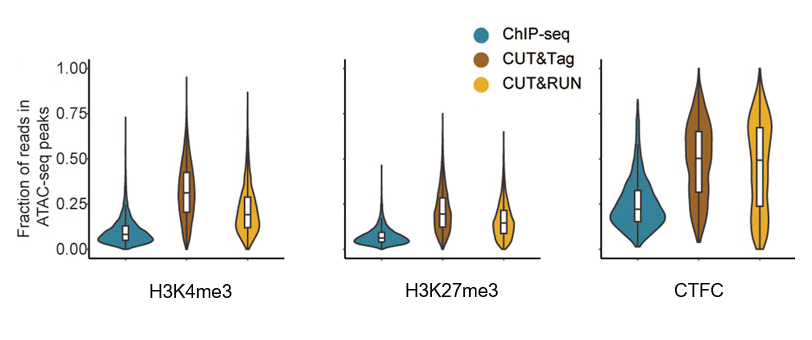
A 2025 benchmark study comparing CUT&RUN, CUT&Tag, and ChIP-seq in haploid round spermatids reported that "signals in CUT&Tag and CUT&RUN were more enriched in accessible chromatin compared to those in ChIP-seq" and concluded that these findings suggest CUT&Tag and CUT&RUN may exhibit bias toward more accessible chromatin. Because this benchmark was performed in a specific biological system, it should not be interpreted as proving the same magnitude of bias across all cell types and targets. However, it provides recent comparative evidence that accessibility-associated enrichment can remain detectable even in optimized workflows. (Wang et al., Frontiers in Cell and Developmental Biology, 2025. Link)
This accessibility-associated signal becomes especially important in negative controls. IgG-only or no-antibody controls can generate recurrent peak patterns that overlap with regions of high genomic accessibility or other problematic genomic regions. This matters because an apparent peak in a CUT&RUN or CUT&Tag dataset may reflect true target binding, accessibility-associated background, recurrent technical artifact, or a combination of these effects.
How the field has responded to open chromatin bias
Importantly, open chromatin bias and recurrent background regions are not hidden issues. Method developers, academic users, and commercial suppliers have recognized that CUT&RUN and CUT&Tag require careful controls and data interpretation. Over time, the field has introduced several practical countermeasures, including IgG or no-antibody negative controls, optimized salt conditions, spike-in normalization, improved peak-calling workflows, ENCODE blacklist filtering, and CUT&RUN-specific suspect-region lists.
The field's response has evolved alongside the methods themselves. CUT&RUN was introduced in 2017 as a targeted nuclease strategy for mapping protein-DNA interactions. CUT&Tag was introduced in 2019 as a Tn5-based targeted tagmentation method for efficient chromatin profiling from small samples and single cells. In 2020, CUTAC demonstrated that related tethered tagmentation chemistry could be adapted specifically for chromatin accessibility mapping, further highlighting the close relationship between these enzymatic workflows and accessible chromatin. (CUT&RUN, eLife 2017; CUT&Tag, Nature Communications 2019; CUTAC, eLife 2020)
By 2023, the problem had become concrete enough that researchers published a dedicated CUT&RUN suspect list of problematic genomic regions. Nordin et al. compiled human and mouse suspect lists from CUT&RUN data, identifying regions consistently called as peaks in negative controls. The authors reported that these regions may persist even after SEACR or MACS2 peak calling against negative controls and after ENCODE blacklist removal. They also experimentally validated the suspect lists using repeated negative-control experiments and reported that the lists captured more than 80% of the peaks identified in those controls. This was a transparent and useful contribution to the field, and it gave researchers a practical tool for improving CUT&RUN data interpretation. (Nordin et al., Genome Biology, 2023. Link)
Commercial CUT&RUN and CUT&Tag suppliers have also attempted to address these issues through optimized wash conditions, high-salt buffers, recommended negative controls, spike-in normalization options, and analysis recommendations. These are meaningful improvements, and for many targets they can produce useful, high-quality datasets. However, most of these measures are mitigation strategies that reduce, reveal, normalize, or correct background rather than fully eliminating its source. They do not fully remove the underlying risk that MNase- or Tn5-based workflows can generate accessibility-associated or recurrent non-target signal under some experimental conditions.
| Field response | What it helps address | Why it may still be insufficient |
|---|---|---|
| IgG or no-antibody controls | Help identify non-specific peaks and background signal. | Controls reveal the problem but do not prevent non-specific chromatin release during the assay. |
| High-salt washes and optimized buffers | Reduce some non-specific binding and improve signal-to-noise. | They may not fully overcome accessibility-associated enzyme behavior, and harsher conditions may affect weak or transient protein-chromatin interactions. |
| ENCODE blacklist and CUT&RUN suspect-list filtering | Remove known problematic genomic regions from downstream analysis. | Filtering improves interpretation but also confirms that recurrent false-positive regions can remain common enough to require post-assay correction. |
| Spike-in normalization | Improves comparison between samples and helps control for technical variation. | Normalization does not distinguish true target enrichment from accessibility-associated or recurrent background signal at specific genomic loci. |
| Computational correction tools | Adjust read distributions and reduce accessibility-associated signal after sequencing. | Correction cannot fully recover information that was biased during the experimental release or tagmentation step. |
Additional context comes from computational correction efforts for CUT&Tag. PATTY was developed specifically to address Tn5-related open chromatin bias in CUT&Tag datasets. The authors reported that Tn5 preference toward accessible chromatin can alter CUT&Tag read distributions and confound downstream analysis, especially in sparse single-cell data. They further reported that open chromatin bias was present across published CUT&Tag datasets, including datasets generated with optimized high-salt protocols. While this work is still in preprint at time of writing, it provides potential and emerging evidence to support the practical concern that improved washing and analysis workflows may reduce, but not necessarily eliminate, assay-level bias. (PATTY bioRxiv preprint; PubMed record)
These concerns also appear in user discussions and troubleshooting forums, where researchers frequently describe high IgG background, ATAC-like peak patterns, and uncertainty around peak interpretation. While such reports are anecdotal, they reflect practical pain points that align with the published technical literature.
Together, these developments show that the field has handled the issue transparently and constructively. The key limitation is that most solutions are control-based, wash-based, filter-based, normalization-based, or computational. They help researchers identify, reduce, normalize, or filter accessibility-associated background, but they do not fully remove the risk of artifact generation at the experimental cleavage or tagmentation stage. For researchers seeing high IgG background, ATAC-like peak patterns, excessive promoter or enhancer signal, or weak discrimination between target and control tracks, a method-level alternative may be more appropriate than adding another layer of correction.
These characteristics carry practical consequences for data interpretation. Active histone marks such as H3K4me3 and H3K27ac, as well as many transcription factors, are frequently associated with promoters, enhancers, and other accessible regulatory elements. In these contexts, true biological enrichment and accessibility-associated background may overlap. For repressive marks, weakly enriched transcription factors, or targets expected to bind outside highly accessible regions, accessibility-associated background can obscure more subtle signals or complicate peak-calling decisions.
For this reason, well-designed CUT&RUN and CUT&Tag studies often benefit from multiple safeguards: matched IgG controls, sufficient biological replicates, careful digestion or tagmentation optimization, spike-in normalization when appropriate, suspect-region filtering, and orthogonal validation by qPCR, ChIP-seq, ATAC-seq, or other methods. These steps help improve confidence, but they also add time, cost, and analytical complexity.
Addressing the issue at the root with CUT&LUNCH
While CUT&RUN and CUT&Tag remain valuable methods when used with appropriate controls and analysis practices, researchers who repeatedly observe high background or accessibility-associated signal may benefit from considering an assay designed to reduce non-specific chromatin release at the experimental stage.
EpigenTek developed CUT&LUNCH (Cleavage Under Target and Liberate Unique Nucleic Complex Homogenously) for this type of challenge. Rather than relying only on post-assay filtering, high-stringency washing, or computational correction, CUT&LUNCH uses an antibody-directed dual-end cleavage approach with a unique enzyme (not MNase or Tn5) designed to selectively release and recover target-associated DNA fragments while reducing non-specific DNA recovery. EpigenTek's product development data shows that CUT&LUNCH provides low non-specific DNA binding of approximately 3-5%, completes the stand-alone assay in less than two hours, does not require ConA immobilization, and supports analysis with existing ChIP-seq bioinformatics pipelines. (CUT&LUNCH overview; CUT&LUNCH Assay Kit)
For sequencing applications, the EpiNext CUT&LUNCH-Seq Kit is designed to move from intact cells to ready-to-use library DNA in under three hours. The workflow avoids nuclei isolation, does not require a secondary antibody, and is designed to generate target-enriched DNA libraries while reducing non-specific protein-DNA recovery. (CUT&LUNCH-Seq Kit)
| Common CUT&RUN/CUT&Tag challenges | Typical mitigation approach | CUT&LUNCH approach |
|---|---|---|
| Non-specific or recurrent control peaks | IgG/no-antibody controls, blacklist filtering, suspect-list filtering. | Designed to eliminate non-specific DNA at the assay level. |
| Open chromatin-associated signal | High-salt conditions, optimized washes, computational correction. | Designed to recover only targeted open chromatin signals via affinity pulldown. |
| Added analysis complexity | Additional controls, filtering steps, spike-ins, and orthogonal validation. | Designed to produce cleaner input for downstream ChIP-seq style analysis pipelines. |
EpigenTek's comparative performance data show that CUT&LUNCH histone marker peak profiles are consistent with ENCODE ChIP-seq profiles and produce fewer false signals than traditional CUT&RUN when tested in MCF-7 cells. For researchers who need high-specificity protein-DNA interaction data, these results support CUT&LUNCH as a practical assay-level option for reducing accessibility-associated background and non-specific cleavage artifacts. (Data summary)
CUT&RUN and CUT&Tag remain useful, widely adopted methods when paired with the right controls and analysis practices, but their known accessibility-associated background makes method selection important. The field has introduced multiple mitigation strategies, including optimized buffers, negative controls, suspect-region filtering, spike-in normalization, and computational correction. However, recent evidence still suggests that these approaches reduce rather than fully eliminate the issue. For laboratories that need cleaner target-specific enrichment with less accessibility-associated background, CUT&LUNCH provides a practical assay-level alternative.
For laboratories troubleshooting CUT&RUN or CUT&Tag background, or for researchers planning experiments where peak specificity is critical, CUT&LUNCH offers a streamlined alternative designed to improve target selectivity, reduce non-specific background, and simplify downstream interpretation.
References and additional reading
- Skene and Henikoff. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. eLife, 2017. View article
- Kaya-Okur et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nature Communications, 2019. View article
- Henikoff et al. Efficient chromatin accessibility mapping in situ by nucleosome-tethered tagmentation. eLife, 2020. View article
- Nordin et al. The CUT&RUN suspect list of problematic regions of the genome. Genome Biology, 2023. View article
- Wang et al. Benchmark comparison of CUT&RUN, CUT&Tag, and ChIP-seq signal enrichment in accessible chromatin. Frontiers in Cell and Developmental Biology, 2025. View article
- Hu et al. PATTY corrects open chromatin bias for improved bulk and single-cell CUT&Tag analysis. bioRxiv preprint, first posted 2025 and revised 2026. View preprint | PubMed record
- EpigenTek CUT&LUNCH overview. View page
- EpiNext CUT&LUNCH Assay Kit. View product
- EpiNext CUT&LUNCH-Seq Kit. View product