 Cart (0)
Cart (0)


Many epitranscriptomics studies begin with a simple observation: total RNA from treated cells, diseased tissue, differentiating cells, viral infection models, or edited cell lines shows a measurable change in N6-methyladenosine, or m6A. That result is useful. It can show that RNA methylation biology is altered, that a perturbation affects the epitranscriptome, or that a writer, reader, or eraser pathway should be investigated.
The next question is harder: which RNAs are responsible for the change?
A bulk m6A result does not identify modified transcripts, modification sites, isoforms, or RNA classes. It also does not distinguish between a true methylation change and a shift in transcript abundance, RNA composition, or sample quality. This is why recent methods-focused reviews frame m6A detection as a workflow decision, not a single-assay decision [1].
A practical m6A study usually moves through three stages: global quantification, transcript-associated enrichment or mapping, and targeted validation. EpigenTek m6A workflow coverage supports that progression, with products for global measurement, m6A RNA enrichment, m6A-seq library preparation, and other antibody-based applications.
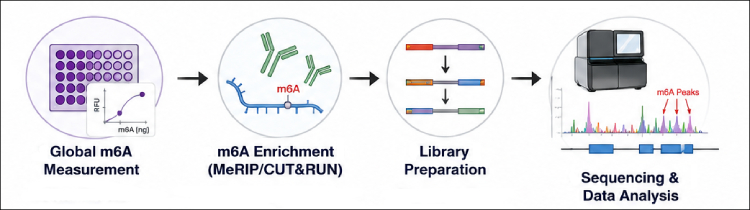
Experimental design rules for moving beyond bulk RNA
A strong m6A study starts with a clear endpoint. Researchers should decide whether they need global change, transcript-associated enrichment, site-level mapping, isoform context, or modification stoichiometry. These endpoints are related, but they are not interchangeable.
- For global experiments, use high-quality RNA, normalize input amount, include positive and negative controls, and run biological replicates. If the biological question is mRNA-specific, consider whether total RNA is the right input or whether mRNA enrichment or rRNA depletion is needed before downstream analysis.
- For enrichment experiments, include input RNA and IgG controls. Analyze enrichment in the context of transcript abundance, because differential expression can mimic or obscure methylation differences. McIntyre et al. highlighted reproducibility limits in detecting m6A peaks and peak changes using MeRIP/m6A-seq, reinforcing the importance of controls and careful analysis [2].
- For sequencing experiments, prioritize biological replication and clean RNA over simply increasing read depth. More reads cannot compensate for degraded RNA, poor controls, or underpowered design. When candidate transcripts emerge, validate them by enrichment-qPCR, orthogonal site-directed methods, or perturbation of relevant writer, eraser, or reader proteins.
Finally, keep assay scope clear. Global m6A quantification is not transcript mapping. m6A enrichment is not necessarily single-nucleotide mapping. m6A detection is not the same as methyltransferase or demethylase activity. RNA methylation is not DNA methylation. m6A is not 5-mC RNA. These distinctions make the difference between a useful m6A result and an overinterpreted one.
What global m6A quantification answers
Global m6A assays ask whether the total m6A content of an RNA preparation differs between conditions. That makes them well suited for screening treated versus untreated samples, comparing time points, evaluating writer or eraser perturbations, and prioritizing samples before deeper mapping.
For global m6A screening, the EpiQuik m6A RNA Methylation Quantification Kit (P-9005) uses an ELISA-based colorimetric readout, while P-9008 provides a fluorometric alternative for labs that prefer fluorescence-based detection. These assays help quantify overall m6A status in total RNA and can guide whether transcript-level enrichment or sequencing is warranted.
The main limitation is interpretive. A global assay does not show whether m6A changed on MYC, SOX2, a viral transcript, a cytokine mRNA, or a specific 3' UTR. It also does not show whether the signal came from mRNA, rRNA, tRNA, snRNA, or another RNA class. Researchers should report the result as global m6A status in the tested RNA preparation, not as evidence of modification at a specific transcript or site.
This distinction is especially important because m6A can influence RNA splicing, nuclear export, translation, decay, localization, and interactions with RNA-binding proteins through context-dependent mechanisms [3,4].
Why transcript-level mapping changes the biological question
The field moved beyond bulk RNA when transcriptome-wide studies showed that m6A is patterned across the transcriptome. Dominissini et al. used m6A-seq to identify more than 12,000 m6A sites in transcripts from more than 7,000 human genes, with enrichment around stop codons and long internal exons [5]. Meyer et al. reported m6A-containing mRNAs from 7,676 mammalian genes, with enrichment near stop codons and in 3' UTRs, and observed tissue-specific regulation including changes during brain development [6].
These studies changed the question from "Did m6A change?" to "Which transcript regions carry m6A, and how does that relate to RNA regulation?"
That shift matters in cancer, stem cell biology, development, neuroscience, immunology, and viral RNA research. A tumor model may show little change in total m6A while redistributing m6A across oncogenic transcripts. A differentiation model may show broad m6A remodeling without a simple increase or decrease in bulk RNA methylation. A viral infection model may alter host and viral RNA modification patterns in different directions.
For this bridge from global signal to transcript-associated analysis, the EpiQuik CUT&RUN MeRIP Kit (P-9018) fits naturally as an enrichment step before qPCR or sequencing.
MeRIP and m6A enrichment: useful, but regional
Methylated RNA immunoprecipitation, often called MeRIP or m6A-RIP, remains one of the most widely used approaches for transcript-associated m6A analysis. In a typical workflow, RNA is fragmented, m6A-containing fragments are enriched with an anti-m6A antibody, and recovered RNA is analyzed by qPCR or sequencing.
The strength of MeRIP-style enrichment is that it can survey many transcripts without requiring prior knowledge of the modified site. The limitation is that standard enrichment identifies m6A-enriched regions, not exact modified adenosines. A peak near a stop codon or within a 3' UTR supports regional m6A enrichment. It does not prove which adenosine is modified, how many transcript molecules are modified, or whether the modification directly causes a functional effect.
Linder et al. developed miCLIP to improve resolution by using antibody-RNA crosslinking signatures to map m6A and m6Am at single-nucleotide resolution [7]. This illustrates a general rule: enrichment-based methods are often practical and scalable, while site-resolution methods usually add complexity.
P-9018 is best positioned for researchers who want m6A-enriched RNA for candidate transcript qPCR or downstream sequencing.
Mapping does not automatically mean stoichiometry
A second common interpretation problem is treating enrichment as modification fraction. A strong m6A peak does not necessarily mean that most copies of a transcript are methylated. It may reflect transcript abundance, antibody recovery, local sequence context, fragment behavior, or the presence of one or more modified residues within a region.
Molinie et al. addressed this issue with m6A-LAIC-seq, designed to estimate m6A levels and characterize isoforms. Their study reported a broad range of nonstoichiometric m6A levels with cell-type specificity and identified differences in alternative polyadenylation between methylated and nonmethylated transcript isoforms [8].
This is a useful reminder for experimental design. A global m6A increase could reflect modest changes across many transcripts, a large change in a smaller transcript subset, altered isoform usage, or changes in RNA abundance. Transcript-level mapping narrows the question, but quantitative interpretation still requires input RNA, expression normalization, biological replication, and validation.
Sequencing options: enrichment, CLIP, chemistry, and direct RNA reads
m6A method choice depends on the question. Global quantification answers whether total m6A changes in a sample. MeRIP or CUT&RUN-style enrichment identifies transcript regions enriched for m6A-containing RNA fragments. CLIP-based methods improve site resolution by using crosslinking-induced mutation or truncation signatures. Chemical and enzyme-assisted methods can provide single-base or quantitative information, depending on the chemistry. Direct RNA sequencing can retain molecule and isoform context, but computational detection remains an active area.
Newer methods continue to expand the toolkit. DART-seq uses an APOBEC1-YTH fusion to induce C-to-U changes near m6A sites and was reported to identify thousands of m6A sites from as little as 10 ng of total RNA [9]. m6A-SAC-seq enables quantitative, whole-transcriptome m6A mapping at single-nucleotide resolution and was reported to require about 30 ng of poly(A) or rRNA-depleted RNA [10]. Nanopore direct RNA sequencing adds single-molecule potential, but a systematic comparison of 10 m6A mapping tools found tradeoffs between precision and recall and showed that negative controls can improve precision by reducing intrinsic bias [11].
For labs moving directly from enrichment to sequencing libraries, the EpiNext CUT&RUN RNA m6A-Seq Kit (P-9016) provides an integrated m6A-seq workflow for Illumina-compatible libraries. It is the more direct fit when NGS, rather than enrichment-qPCR, is the planned endpoint.
Antibody choice still matters
Many m6A workflows depend on antibody recognition. This makes antibody choice a core design variable, not a minor reagent decision. Antibody performance can affect enrichment, background, apparent peak distribution, and reproducibility.
EpigenTek’s A-1801 is the polyclonal option commonly aligned with MeRIP and related enrichment workflows, including P-9018 and P-9016 examples. A-1802 provides a monoclonal option for ELISA, dot blot, IF, MeRIP, and nucleotide array applications.
Controls are essential. At minimum, m6A enrichment experiments should include input RNA for normalization, an IgG or non-immune control, biological replicates, and candidate validation. For unfamiliar sample types, optimization should be expected.
Product selector: matching the assay to the m6A question
| Research question | Relevant product | Product fit |
|---|---|---|
| Did total m6A change between RNA samples? | EpiQuik m6A RNA Methylation Quantification Kit (Colorimetric) (#P-9005) | Global m6A screening from total RNA; 100 to 300 ng input per assay, with controls for relative or absolute quantification. |
| Do I need a fluorometric global m6A readout? | EpiQuik m6A RNA Methylation Quantification Kit (Fluorometric) (#P-9008) | Global m6A quantification when fluorescence detection is preferred. |
| Which RNA regions are enriched for m6A? | EpiQuik CUT&RUN m6A RNA Enrichment (MeRIP) Kit (#P-9018) | m6A-containing RNA enrichment before qPCR or NGS; includes non-immune IgG and m6A positive control workflow elements. |
| Do I need enrichment plus NGS library preparation? | EpiNext CUT&RUN RNA m6A-Seq Kit (#P-9016) | Integrated enrichment-to-library workflow for Illumina-compatible m6A-seq. |
| Do I need a polyclonal anti-m6A antibody? | N6-methyladenosine (m6A) Polyclonal Antibody (#A-1801) | Antibody-based m6A detection or enrichment workflows, including MeRIP-oriented applications. |
| Do I need a monoclonal anti-m6A antibody? | N6-methyladenosine (m6A) Monoclonal Antibody [2H6] (#A-1802) | Monoclonal antibody option for ELISA, dot blot, IF, MeRIP, and nucleotide array applications. |
Global m6A quantification is often the right first experiment, but it should be treated as a screen and prioritization tool. To connect RNA modification changes to mechanism, researchers need transcript-associated enrichment, sequencing, expression-aware analysis, and targeted validation. A staged workflow using global quantification, m6A RNA enrichment, m6A-seq, and antibody-supported validation helps move the study from "m6A changed" to "these transcript regions are associated with m6A under these conditions."
References
- Moshitch-Moshkovitz S, Sevilla-Sharon M, Ashwal-Fluss R, Glick-Saar E, Rechavi G, Dominissini D. mRNA m6A detection. Nat Rev Methods Primers. 2024;4(1):87. doi:10.1038/s43586-024-00365-9. View article
- McIntyre ABR, Gokhale NS, Cerchietti L, Jaffrey SR, Horner SM, Mason CE. Limits in the detection of m6A changes using MeRIP/m6A-seq. Sci Rep. 2020;10(1):6590. Published 2020 Apr 20. doi:10.1038/s41598-020-63355-3 View article
- Roundtree IA, Evans ME, Pan T, He C. Dynamic RNA Modifications in Gene Expression Regulation. Cell. 2017;169(7):1187-1200. doi:10.1016/j.cell.2017.05.045 View article
- Zaccara S, Ries RJ, Jaffrey SR. Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol. 2019;20(10):608-624. doi:10.1038/s41580-019-0168-5 View article
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485(7397):201-206. Published 2012 Apr 29. doi:10.1038/nature11112 View article
- Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3' UTRs and near stop codons. Cell. 2012;149(7):1635-1646. doi:10.1016/j.cell.2012.05.003 View article
- Linder B, Grozhik AV, Olarerin-George AO, Meydan C, Mason CE, Jaffrey SR. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods. 2015;12(8):767-772. doi:10.1038/nmeth.3453 View article
- Molinie B, Wang J, Lim KS, et al. m(6)A-LAIC-seq reveals the census and complexity of the m(6)A epitranscriptome. Nat Methods. 2016;13(8):692-698. doi:10.1038/nmeth.3898 View article
- Meyer KD. DART-seq: an antibody-free method for global m6A detection. Nat Methods. 2019;16(12):1275-1280. doi:10.1038/s41592-019-0570-0 View article
- Hu L, Liu S, Peng Y, et al. m6A RNA modifications are measured at single-base resolution across the mammalian transcriptome. Nat Biotechnol. 2022;40(8):1210-1219. doi:10.1038/s41587-022-01243-z View article
- Zhong ZD, Xie YY, Chen HX, et al. Systematic comparison of tools used for m6A mapping from nanopore direct RNA sequencing. Nat Commun. 2023;14(1):1906. Published 2023 Apr 5. doi:10.1038/s41467-023-37596-5 View article