 Cart (0)
Cart (0)


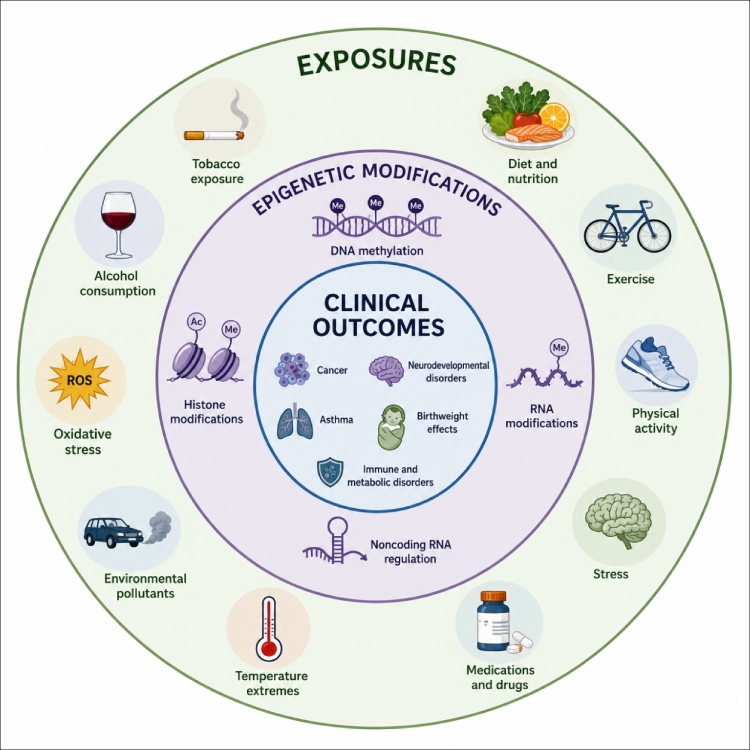
The exposome is the full set of environmental exposures a person experiences across life, including chemical, physical, nutritional, behavioral, social, and biological factors. The concept was introduced to help environmental health researchers complement genomic information with better exposure measurement across the life course [1]. For today’s exposome studies, the challenge is not simply asking whether the environment affects biology. The harder question is how to measure molecular responses to complex, overlapping exposures in a way that is interpretable, reproducible, and useful for future research.
DNA methylation is one of the most widely studied molecular readouts for this challenge. It is chemically stable enough to measure across many biological specimens, closely connected to gene regulation, and compatible with both focused assays and genome-wide profiling. In environmental health, toxicology, stress biology, and population epigenetics, DNA methylation can help connect exposure history to cellular response, including prenatal and early-life exposure effects that may shape later biological risk [2,3].
However, DNA methylation data should be interpreted carefully. A methylation signal is not automatically a causal mechanism, a diagnostic marker, or proof of exposure. It may reflect exposure, tissue composition, aging, inflammation, disease state, genetic background, or several of these factors at once. The practical goal is to match the assay to the research question.
EpigenTek supports this decision process with workflows for global 5-mC and 5-hmC measurement, bisulfite-based downstream analysis, and reduced representation bisulfite sequencing. For exposome studies, this creates a practical path from broad screening to targeted validation to genome-wide discovery.
How researchers should connect assay choice to study stage
- For early exploratory work, use global 5-mC and 5-hmC assays to compare exposure groups and determine whether a broader methylation response is present. This can help prioritize samples and conditions before moving into sequencing.
- For hypothesis-driven work, use bisulfite conversion and downstream locus-specific analysis to test candidate genes, CpG islands, enhancers, or regulatory regions. This is useful when prior literature, toxicology pathways, or pilot sequencing data point to specific regions.
- For discovery work, use RRBS when the goal is to identify CpG-rich regions associated with an exposure, treatment, or stress condition. Follow discovery results with targeted validation and functional interpretation.
This staged approach helps avoid two common problems: running sequencing too early without a clear biological question, or relying only on global methylation when locus-level information is needed.
Why the exposome needs measurable molecular signals
Environmental exposures rarely occur one at a time. Air pollution, diet, metals, endocrine-active compounds, psychosocial stress, smoking, medication, infection, circadian disruption, and the built environment can overlap across development and aging. These exposures may act through oxidative stress, immune activation, endocrine signaling, metabolic stress, mitochondrial changes, or altered chromatin regulation [3].
DNA methylation is attractive because it can act as both a biological readout and a hypothesis-generating signal. At the molecular level, DNA methylation most often refers to 5-methylcytosine, or 5-mC, formed by DNA methyltransferases at cytosine bases. In mammals, 5-mC occurs mainly at CpG sites and is involved in gene regulation, genomic imprinting, transposon control, development, and cell identity [4]. A related mark, 5-hydroxymethylcytosine, or 5-hmC, is generated through TET-mediated oxidation of 5-mC and is especially relevant in development, neuronal biology, stem cell biology, and active DNA demethylation pathways.
This distinction matters in environmental epigenetics. A decrease in global 5-mC, an increase in methylation at a promoter, and a change in 5-hmC are not equivalent findings. They may point to different biology and require different assays.
What environmental stress can do to DNA methylation
Environmental stress can influence DNA methylation through several routes. Some exposures affect one-carbon metabolism and methyl donor availability, which can influence the biochemical supply of methyl groups. Other exposures generate oxidative stress or inflammatory signaling, which can alter DNA methyltransferase activity, TET activity, repair pathways, and cell composition [3]. Chronic stressors may also influence endocrine and immune pathways that reshape methylation patterns in blood or tissue.
Large population studies show why DNA methylation has become a central tool in exposure biology. Smoking is one of the clearest examples. A large blood-based meta-analysis identified thousands of smoking-associated CpG sites, including signals in genes connected with xenobiotic response and inflammation [5]. Maternal smoking during pregnancy has also been associated with widespread CpG methylation differences in newborn blood across a large multi-cohort analysis [6]. These findings show that some exposures can produce strong and reproducible methylation signatures.
Other exposures are more complex. Air pollution, greenness, urbanicity, and psychosocial stress can show methylation associations, but effect sizes, tissue relevance, and replication may vary. A recent urban exposome study linked urban environmental factors with blood DNA methylation differences across life stages and reported stronger associations in young children and adults, while also noting that exposure-specific methylome signatures can be difficult to separate [7]. Prenatal stress meta-analysis data also suggest that stressful life events during pregnancy can be associated with methylation differences at selected loci, although these signals are often more modest than smoking-associated signatures [8].
The lesson for researchers is clear: DNA methylation is useful for exposome research, but study design determines whether the results are interpretable.
Start with the right level of methylation measurement
A strong exposome study often uses a tiered design. The first tier asks whether exposure groups show a broad shift in DNA modification. The second asks whether candidate genes or regions are affected. The third asks where genome-wide methylation differences occur.
Global 5-mC and 5-hmC screening
Global methylation assays are useful when the first question is broad: does an exposure, treatment, stress condition, or sample group show an overall change in 5-mC or 5-hmC content?
For this screening step, the MethylFlash Global DNA Methylation (5-mC) ELISA Easy Kit (P-1030) provides a practical way to quantify global 5-mC from purified DNA. It is especially relevant for comparing exposure groups before committing to deeper sequencing. In parallel, the MethylFlash Global DNA Hydroxymethylation (5-hmC) ELISA Easy Kit (P-1032) helps researchers evaluate whether environmental stress is associated with changes in the oxidized cytosine mark 5-hmC.
This 5-mC and 5-hmC pairing is useful because a global 5-mC-only workflow may miss biologically meaningful shifts in DNA demethylation biology. For example, a condition that changes TET activity or oxidative stress may alter 5-hmC without producing a simple global 5-mC pattern. Measuring both marks gives researchers a broader starting view of methylation dynamics.
Global assays do not identify the genomic regions that changed. They are best used as screening or comparison tools, not as a substitute for locus-specific or genome-wide mapping.
Candidate-region and locus-specific methylation analysis
When the study has a focused hypothesis, bisulfite-based methods remain useful. For example, a toxicology group may want to examine methylation in promoters linked to detoxification, inflammation, endocrine signaling, or oxidative stress. A stress biology study may focus on regions related to glucocorticoid signaling, immune response, or neuronal function. A population epigenetics study may validate CpGs identified by an earlier EWAS.
Bisulfite conversion supports this kind of locus-specific analysis by converting unmethylated cytosines while leaving 5-mC protected from conversion, enabling downstream methylation-specific PCR, methylation-sensitive high resolution melting, pyrosequencing, microarray analysis, or bisulfite sequencing [4,9]. The BisulFlash DNA Modification Kit (P-1026) fits this stage by preparing converted DNA for multiple downstream methylation analysis methods.
For exposome studies, the critical issue is DNA quality. Environmental samples may come from archived tissue, blood, plasma or serum, FFPE material, or low-input specimens. Researchers should control for DNA integrity, conversion efficiency, target amplicon length, and batch effects. Poor conversion or degraded DNA can create false methylation patterns that may be mistaken for biological exposure effects.
Genome-scale discovery with RRBS
When researchers do not know which loci are affected, sequencing-based methylation profiling is better suited than a global methylation assay. Whole-genome bisulfite sequencing, or WGBS, is the most comprehensive bisulfite-based approach because it can profile methylation across the genome at single-base resolution. However, WGBS can require greater sequencing depth, higher analysis burden, and larger budgets than many screening or pilot studies allow.
Reduced representation bisulfite sequencing, or RRBS, offers a more focused alternative. Rather than surveying nearly all genomic cytosines, RRBS enriches a reduced portion of the genome that is relatively CpG-rich, providing single-base methylation information across many promoters, CpG islands, and other CpG-dense regions [4]. For exposome studies, this makes RRBS useful when researchers need more regional detail than a global 5-mC or 5-hmC assay can provide, but do not need the full breadth of WGBS.
The EpiNext RRBS Library Fast Kit (P-1069) supports RRBS library preparation for Illumina sequencing. In exposome projects, RRBS can help identify candidate exposure-responsive regions, prioritize CpGs for validation, and compare methylation patterns across treatment groups, cohorts, or time points.
The strength of RRBS is efficient, CpG-enriched methylation discovery across a defined representation of the genome. It is strongest when the research question is compatible with CpG-rich region enrichment and when sequencing depth, sample size, and bioinformatics analysis are planned in advance.
Designing better exposome methylation studies
The biggest risk in environmental epigenetics is overinterpreting methylation changes without enough context. Strong study design should address at least six issues.
1. Sample type and tissue relevance
DNA methylation is tissue- and cell-type-specific. Blood is practical for population studies, but blood methylation can reflect immune cell composition as much as exposure biology. Brain, placenta, liver, lung, germ cells, tumor tissue, and epithelial samples may answer different questions. When the exposure is systemic, blood may be informative. When the exposure affects a specific organ, tissue relevance becomes more important.
2. Timing of exposure
The same exposure may have different methylation effects depending on whether it occurs during prenatal development, childhood, adulthood, or aging. Developmental windows can be especially sensitive because DNA methylation is actively remodeled during early life, making prenatal and early-life exposure timing especially important in exposome study design [2,6,8]. Longitudinal sampling, when feasible, is stronger than a single time point [6,8].
3. Exposure measurement
A methylation study is only as strong as its exposure data. Personal monitoring, validated questionnaires, geographic exposure models, biomarker measurements, and repeated exposure assessments all improve interpretation. Exposome research often benefits from combining external exposure data with internal molecular data [1,7].
4. Global versus locus-specific interpretation
A global 5-mC shift suggests broad methylation change but does not reveal where it occurs. A promoter methylation change may affect gene regulation but does not represent the whole methylome. RRBS or EWAS can identify loci, but discovery results usually need validation in independent samples or targeted assays.
5. 5-mC versus 5-hmC
Bisulfite-based methylation analysis can be limited in distinguishing 5-mC from 5-hmC unless additional chemistry or dedicated assays are used. In tissues or conditions where 5-hmC is biologically important, researchers should consider measuring 5-hmC directly rather than assuming all methylation signal reflects 5-mC.
6. Confounders and covariates
Age, sex, ancestry, smoking, medication, disease status, diet, BMI, cell composition, and batch effects can influence methylation data. Environmental epigenetics studies should include appropriate covariates, negative controls, and replication plans. This is especially important in exposome studies, where multiple correlated exposures can make it difficult to assign methylation differences to one specific stressor [7].
| Product selector for exposome and DNA methylation workflows | ||
|---|---|---|
| Research goal | Relevant product | Best fit |
| Screen global 5-mC changes | MethylFlash Global DNA Methylation (5-mC) ELISA Easy Kit (P-1030) | Global 5-mC quantification from purified DNA. Useful for comparing exposure groups before locus-specific or sequencing studies. |
| Screen global 5-hmC changes | MethylFlash Global DNA Hydroxymethylation (5-hmC) ELISA Easy Kit (P-1032) | Global 5-hmC quantification from purified DNA. Useful when TET activity, hydroxymethylation, or demethylation biology is relevant. |
| Prepare DNA for targeted bisulfite analysis | BisulFlash DNA Modification Kit (P-1026) | Bisulfite-converted DNA for MS-PCR, MS-qPCR, MS-HRM, pyrosequencing, methylation microarray, or bisulfite sequencing. |
| Discover CpG-rich methylation patterns | EpiNext RRBS Library Fast Kit (P-1069) | RRBS library preparation for Illumina sequencing. Useful for regional methylation discovery across CpG-rich genomic regions. |
The exposome challenges researchers to study mixtures of environmental stressors across time, tissues, and biological pathways. DNA methylation provides one of the most practical molecular entry points into that complexity, but the value of the data depends on choosing the right measurement level. Global 5-mC and 5-hmC assays can show broad methylation shifts. Bisulfite-based workflows can test candidate regions. RRBS can support discovery across CpG-rich regions. When these methods are used strategically, DNA methylation becomes more than a list of altered CpG sites. It becomes a framework for asking better questions about how environmental stress is recorded, regulated, and interpreted in the epigenome.